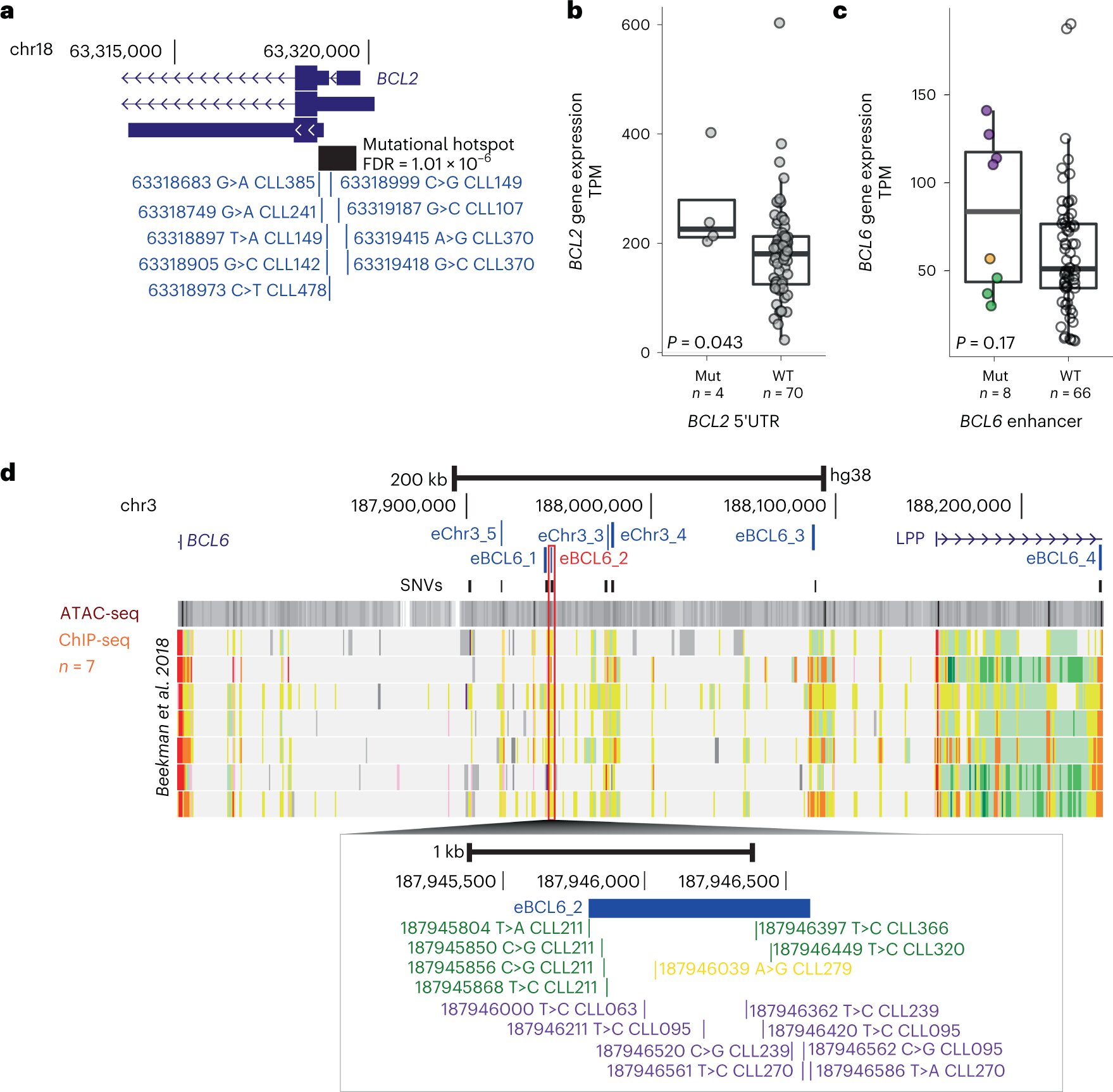
Совместное исследование, проведенное Оксфордским университетом в рамках проекта 100 000 геномов Великобритании, опубликованного сегодня в Nature Genetics, определило пять новых подгрупп наиболее распространенного типа рака крови - хронического лимфолейкоза (ХЛЛ) и связало их с клиническими исходами. Этот новый метод стратификации риска пациентов может привести к более персонализированному уходу за пациентами.
Это первое исследование, в котором анализируются все соответствующие изменения в ДНК по всему геному рака, а не по целевым областям, чтобы классифицировать пациентов с раком и связать эти подгруппы с клиническими исходами. Профессор Анна Шух, кафедра онкологии Оксфордского университета, которая руководила исследованием, сказала: "Мы знаем, что рак в основном является заболеванием, вызываемым изменениями в ДНК, которые приобретаются в течение жизни человека. Лабораторные инструменты, которые мы в настоящее время используем для прогнозирования того, ответит ли пациент на ту или иную терапию, обычно фокусируются на единичных аномалиях в ДНК рака и не позволяют точно предсказать клинический исход пациента. Вот почему мы задали простой вопрос: можем ли мы повысить точность текущего тестирования, изучив все приобретенные изменения ДНК при раке сразу?" В этом исследовании были проанализированы полные последовательности генома 485 пациентов с ХЛЛ, которые были включены в национальные клинические испытания, проводимые университетами Ливерпуля и Лидса, предоставили образцы для британского биобанка ХЛЛ в Ливерпуле и дали согласие на использование их образцов в проекте 100 000 геномов, проводимом Genomics England. Сравнивая данные секвенирования всего генома из раковых и здоровых тканей у этих пациентов, команда смогла отобразить известные и вновь выявленные изменения ДНК, структурные изменения, сигнатуры раковых мутаций и другие глобальные показатели, связанные с ХЛЛ по всему геному. Они идентифицировали 186 различных и повторяющихся геномных изменений и использовали их для определения пяти геномных подгрупп ХЛЛ, которые ассоциируются с различными клиническими исходами. После дополнительной валидации у пациентов, получающих более широкий спектр лечения, чем те, которые включены в это исследование, особенно таргетную терапию, эти новые геномные подгруппы для ХЛЛ могут быть использованы для лучшего руководства выбором методов лечения для улучшения результатов лечения пациентов. Это исследование открывает путь для рутинного клинического применения анализа всего генома для стратификации риска при других типах рака. Анализ команды также выявил 148 новых предполагаемых генетических факторов ХЛЛ. Будущие исследования этих факторов могут раскрыть новые механизмы возникновения и прогрессирования ХЛЛ, что может привести к разработке новых терапевтических средств. Профессор сэр Марк Колфилд, заместитель директора по здравоохранению Лондонского университета Королевы Марии и бывший главный научный сотрудник и руководитель проекта "100 000 геномов" в Genomics England, сказал: "Это крупнейший международный геномный анализ этого вида рака крови, который захватывающе демонстрирует реальную ценность секвенирования всего генома в рамках проекта "100 000 геномов". В нем также используются высококачественные образцы рака крови из Ливерпульского биобанка хронического лимфолейкоза и соответствующие клинические данные, собранные подразделениями клинических исследований в университетах Ливерпуля и Лидса в рамках многоцентровых клинических испытаний, проводимых при поддержке Национального института исследований рака. "Наша работа показывает, что весь геном превосходит классификацию пациентов по группам по сравнению с традиционными целевыми подходами и что мы можем более точно предсказать реакцию на лечение у большего числа пациентов". | |
| Просмотров: 191 | |